High-throughput single-cell transcriptome studies have thrived in animal and human research in recent years, but only a handful of studies have been published in plants. A major reason for this is that plant cells are confined by cell walls, and protoplasting is required to release individual cells. But generating protoplasts remains to be difficult in many tissues and may influence the transcriptome. Therefore, a protoplasting-free method is urgently needed to broaden the application of large-scale single-cell analysis in plants.
Associate Professor Jixian Zhai’s team from the Institute of Plant and Food Science of the Department of Biology at the Southern University of Science and Technology (SUSTech) previously characterized full-length nascent RNAs in Arabidopsis and found a large number of polyadenylated mRNAs that are tightly associated with chromatin. Therefore, they assume that the polyadenylated RNAs in a nucleus may be sufficient to convey information on cell identity.
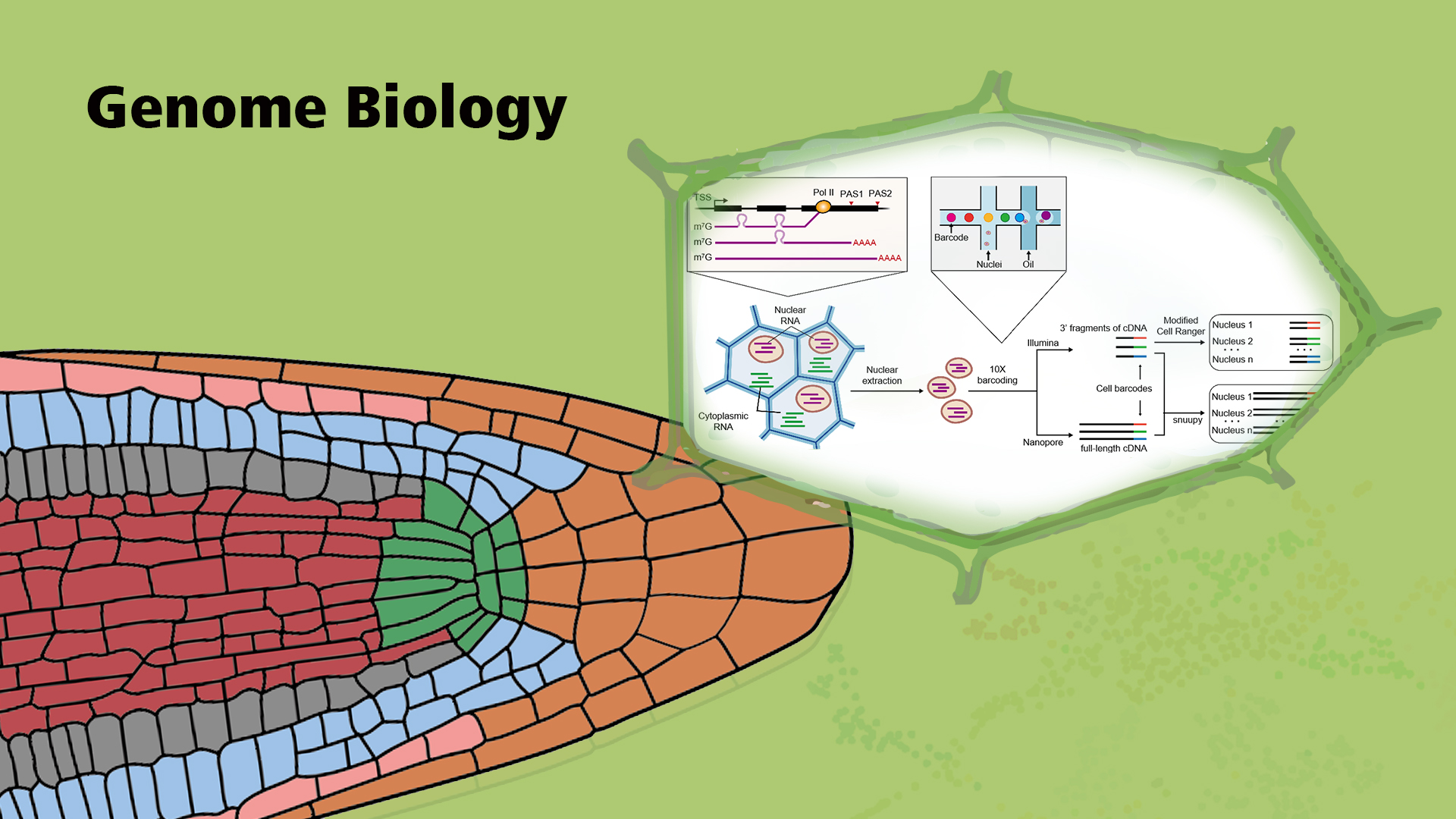
To take full advantage of these intron-containing mRNAs in the nucleus and remove the requirement for protoplasting, Jixian Zhai’s team developed FlsnRNA-seq (protoplasting-free full-length single-nucleus RNA profiling technology), a long-read single-nucleus RNA profiling method in plants by combining 10x Genomics and Nanopore sequencing technologies. This method enables researchers to profile the full-length nuclear mRNA and characterize the status of alternative splicing as well as alternative polyadenylation at the single-cell resolution, and use these extra layers of information to further improve cell-type classification (Figure 1). The study has been published in the journal of Genome biology (IF = 10.806) on February 19, 2021, and entitled “FlsnRNA-seq: protoplasting-free full-length single-nucleus RNA profiling in plants.”

Figure 1. Schematic diagram of FlsnRNA-seq. The purified nuclei are isolated from plant tissue and subjected to library construction according to the 10x Genomics protocol. After reverse transcription and cDNA amplification, half of the cDNA template was used to construct the Illumina library, the other half of the template was used to make the Nanopore library.
First, the authors chose to use the Arabidopsis root to validate the effectiveness of FlsnRNA-seq. They identified 14 cell clusters by analyzing Illumina data. After cell-type annotation, 10 major root cell types were identified (Figure 2). In addition, they found that the dataset generated by FlsnRNA-seq closely resembles the published protoplasting-based single-cell dataset. It indicates that nuclei can be used as a reliable alternative to protoplasts for single-cell RNA sequencing. Furthermore, FlsnRNA-seq facilitates cell type classification and characterization combined with the Nanopore-based full-length RNA sequencing method.

Figure 2. FlsnRNA-seq reveals the diverse cell types in the Arabidopsis root. UMAP visualization of the various cell types clustered using Illumina single-nucleus data.
To validate the robustness of FlsnRNA-seq, the authors applied this method to the multinucleate endosperm isolated from the heart-stage ovules of Arabidopsis, and obtained six clusters covering all previously reported major endosperm cell types. Combining with the Nanopore dataset, they found a cluster dominated by micropylar endosperm exhibits a distinct high ratio of incompletely spliced transcripts (Figure 3). Taken together, the FlsnRNA-seq facilitates cell type classification and characterization by providing extra layers of information in addition to abundance. It would enable plant researchers to explore numerous fascinating plant tissues at the single-cell level and raise more interesting biological questions.
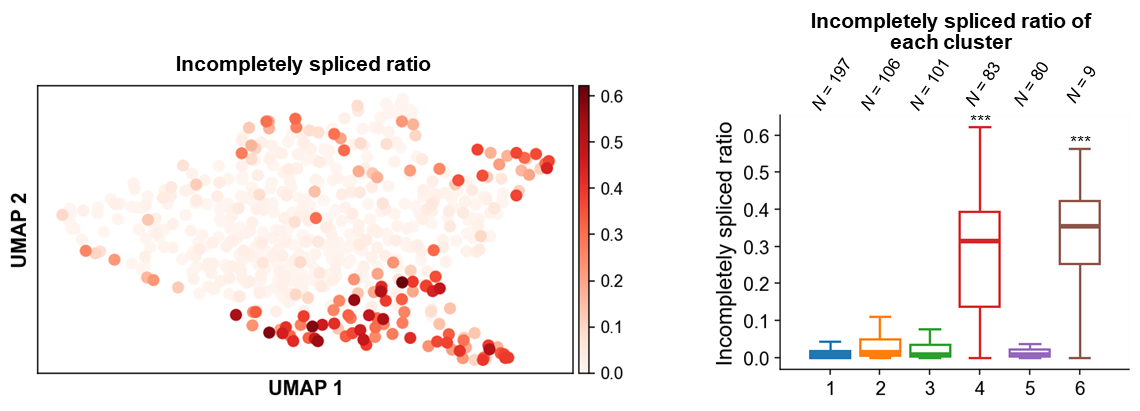
Figure 3. FlsnRNA-seq captures the variation in intron retention levels of different clusters in Arabidopsis endosperm. UMAP visualization of incompletely spliced ratio calculated by Nanopore full-length reads (left panel). Boxplot visualization of the incompletely spliced ratio of each cluster (right panel).
Associate Professor Jixian Zhai is the corresponding author of the paper. The co-first authors are Research Assistant Professors Dr. Yanping Long and Dr. Jinbu Jia, and Ph.D. student Mr. Zhijian Liu. Professor Dr. Wei Chen and Research Associate Professor Dr. Liang Fang also made important contributions to the research.
The research paper received support from the National Key R&D Program of China (NKPs), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams, the Shenzhen Sci-Tech Fund, and the Key Laboratory of Molecular Design for Plant Cell Factory of Guangdong Higher Education Institutes (SUSTech).
Jixian Zhai’s group website link: http://www.jixianzhai.org/
Paper link: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02288-0
Proofread ByAdrian Cremin, Yingying XIA
Photo By






