Advanced Science and Technology at SUSTech” Series Report – Part 2
Gene regulation is one of the central themes of modern molecular biology. In order to understand the organismal development and function, it is essential to reveal the regulatory mechanisms underlying the diverse spatial-temporal gene expression patterns.
Research on post-transcriptional gene regulation is moving into the central stage
Gene expression is a multi-step process. After transcription, mRNAs have to go through a series of intertwining processes to be finally translated into functional proteins. The previous studies clearly showed that transcription alone can explain at most half of the cellular protein abundance. The post‐transcriptional regulation provides cells an extended option to fine‐tune their proteomes. To meet the demands of complex organism development and appropriately respond to environmental stimuli, all these steps need to be finely regulated and the dysregulation could lead to pathological conditions. Compared to the more extensively studied transcriptional regulation, although with a similar, if not higher functional importance, the post-transcriptional gene regulation has remained largely underexplored. Only recently, with its importance being more and more appreciated, research on post-transcriptional processes starts to flourish.
In the recent years, Prof. Wei Chen from the Department of Biology of SUSTech, has been mainly studying the regulation of gene expression, particularly the different processes of post-transcriptional gene regulation.
Quantitative model on mammalian gene expression control
In the last years, the research team of Prof. Chen has developed a series of high-throughput omics technology-based methods to quantitatively analyze the different processes of gene regulation. In 2011, together with Selbach and Wolf labs, they have for the first time measured in mammalian cells for over 6000 genes the mRNA and protein abundance as well as their turnover rates. Based on these genome-wide data, they constructed the first mathematical model to quantitatively describe the mammalian gene regulation at a global level. Their results revealed that the cellular protein abundance is largely determined after RNA transcription (Figure 1).
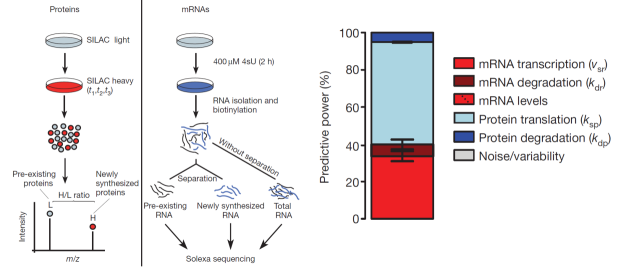
Figure 1. Genome-wide measurement of mRNA and protein abundance as well as their turnover rates in mammalian cells. The data quantitatively described the relative contribution of different regulatory processes to the final cellular protein abundance.
Reconstruction of gene regulation network facilitates precision diagnosis
Post-transcriptional gene regulation includes many different processes, such as RNA splicing, RNA polyadenylation, RNA decay and mRNA translation. Although via various mechanisms, the regulation of these different post-transcriptional processes is in general mediated by the interaction of diffusible trans-regulatory factors (e.g. RBPs, non-coding RNAs) andcis-regulatory elements, which reside mostly in the non-coding regions of mRNAs. To fully understand the regulatory network, the first step is to globally characterize cis– or trans– regulatory factors. Currently, the most efficient strategy to study the cis-elements in a genome-wide manner is to compare the allele-specific gene regulation in a F1 hybrid model system. In F1 hybrids, the nascent RNA transcripts from both parental alleles are subject to the same trans‐regulatory environments; thus, the observed differences in allele‐specific patterns should only reflect the impact of cis‐regulatory divergence. This approach has been successfully used for studying cis‐regulation in RNA transcription. However, in the previous mammalian systems, due to the limited genome divergence between the parental strains, the RNA-seq method could not be used to efficiently distinguish the post-transcriptional regulatory events between the two parental alleles. Therefore, the allele-specific post-transcriptional gene regulation remains largely unexplored in mammals. To overcome this, Chen Lab has developed a new F1 hybrid systems between Mus musculus C57BL/6J and Mus spretus SPRET/EiJ inbred mouse strains. The two parental strains diverged ~1.5 million years (Ma) ago, which resulted in about 35.4 million single nucleotide variants (SNVs) and 4.5 million insertion and deletions (indels) between their genome sequences. Such a high sequence divergence on one hand provides a large number of RNA transcripts harboring potential cis-regulatory variants, on the other hand enables the use of sequencing approach to distinguish allelic RNA transcripts.
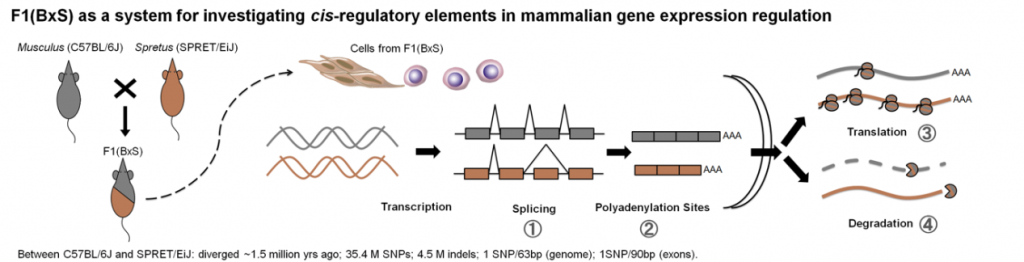
Figure 2. The F1 Hybrid Mouse used to analyze the allele-specific gene regulation.
Based on this hybrid system, Chen Lab has developed a suite of experimental and computational pipelines to quantify the allele-specific gene regulation, including RNA alternative splicing, RNA alternative polyadenylation, RNA decay and mRNA translation (Figure 2). Their studies not only revealed in mammals predominant contribution of cis‐regulatory changes in the evolution of alternative splicing as well as alternative polyadenylation, but also demonstrated that the allele-specific events could be used to identify potential cis-regulatory elements. Now, they are using the same hybrid system to further explore the allele-specific gene regulation across different tissues and under different pathophysiological conditions. Based on these patterns, novel cis-elements and accompanying trans-factors will be discovered and thereafter the regulatory networks functioning in distinct tissues and under different conditions will be reconstructed. The findings of these regulatory networks could be used to analyze cancer genome sequencing data and screen for the driver mutations with impacts in gene dysregulation. All these will facilitate the further understanding of the role of gene regulation in the tumorigenesis and pave the way for the future development of precision medicine in treating cancer patients.
Lab website: http://bio.sustc.edu.cn/chenlab/
Proofread By
Photo By






