Scaffolding is crucial for constructing most chromosome-level genomes. High-throughput chromatin conformation capture (Hi-C) technology has become the primary scaffolding strategy due to its convenience and cost-effectiveness. As sequencing technologies and assembly algorithms advance, constructing haplotype-resolved genomes is increasingly preferred because haplotypes can provide additional genetic information on allelic and non-allelic variations. This information is essential for research such as allele-specific gene expression studies.
ALLHiC is a widely used allele-aware scaffolding tool specifically designed for this purpose. However, this method requires a chromosome-level reference genome from a closely related species, which may not be available for many species. Although it is feasible to assemble and annotate a haplotype-collapsed genome as the reference, this approach noticeably increases the time and cost of genome research. In addition, ALLHiC has been observed to introduce chromosome assignment errors when using the reference genome. These limitations and drawbacks have somewhat hindered the construction of haplotype-resolved genomes, especially in autopolyploids.
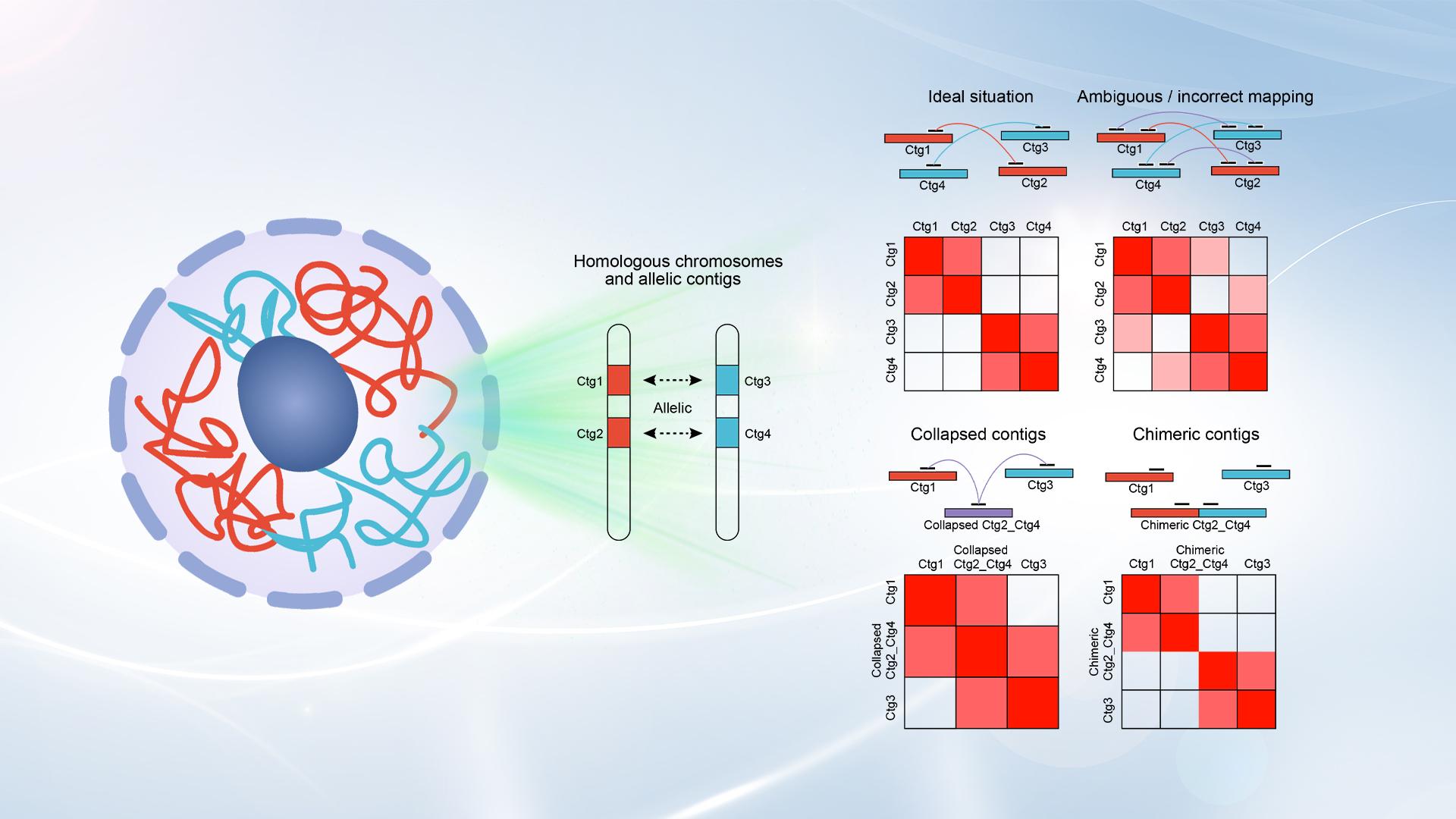
To address this problem, Associate Professor Guoan Chen’s research team from the Department of Human Cell Biology and Genetics, School of Medicine at the Southern University of Science and Technology (SUSTech) has developed a new Hi-C scaffolding tool named HapHiC. This tool enables the scaffolding of chromosome-level haplotypes using Hi-C data without reliance on reference genomes (de novo). They achieved this by introducing a series of algorithmic innovations to identify and handle unique chromatin interaction patterns on misassembled contigs and allelic contigs. Furthermore, this study provides new insights into the challenges in scaffolding haplotype-resolved assemblies by conducting comprehensive analyses on various adverse factors.
Their research was recently published in Nature Plants, entitled “Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes”. Upon the editor’s invitation, the research team also wrote a Research Briefing titled “Achieving de novo scaffolding of chromosome-level haplotypes using Hi-C data” in the same issue. The work received high praise from the journal editor and experts.
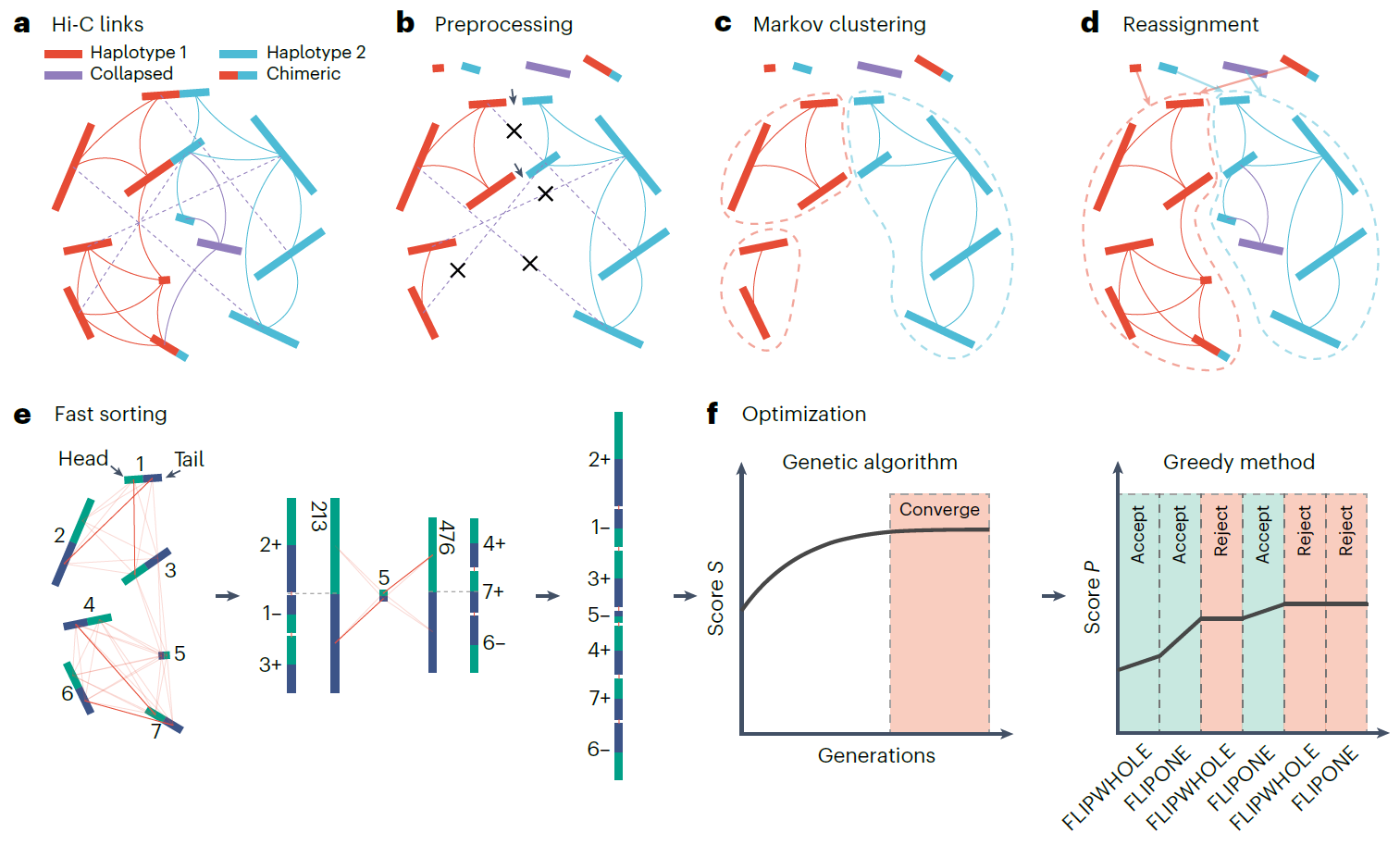
Figure 1. Overview of the HapHiC pipeline
The research team simulated various adverse factors that could impede the scaffolding of haplotype-resolved assemblies based on an autotetraploid genome. A comprehensive performance evaluation showed that HapHiC exhibits higher tolerance to different types of assembly errors compared to other widely used Hi-C scaffolding tools, as reflected by higher scaffold contiguities and lower misassignment rates. After chromosome assignment, HapHiC orders and orients contigs within each chromosome. The iterative scaffolding algorithm 3D-DNA and optimization algorithm in ALLHiC were improved and combined in HapHiC, resulting in more accurate and efficient ordering and orientation of contigs. Especially when contigs are short, HapHiC significantly outperforms other scaffolding tools.
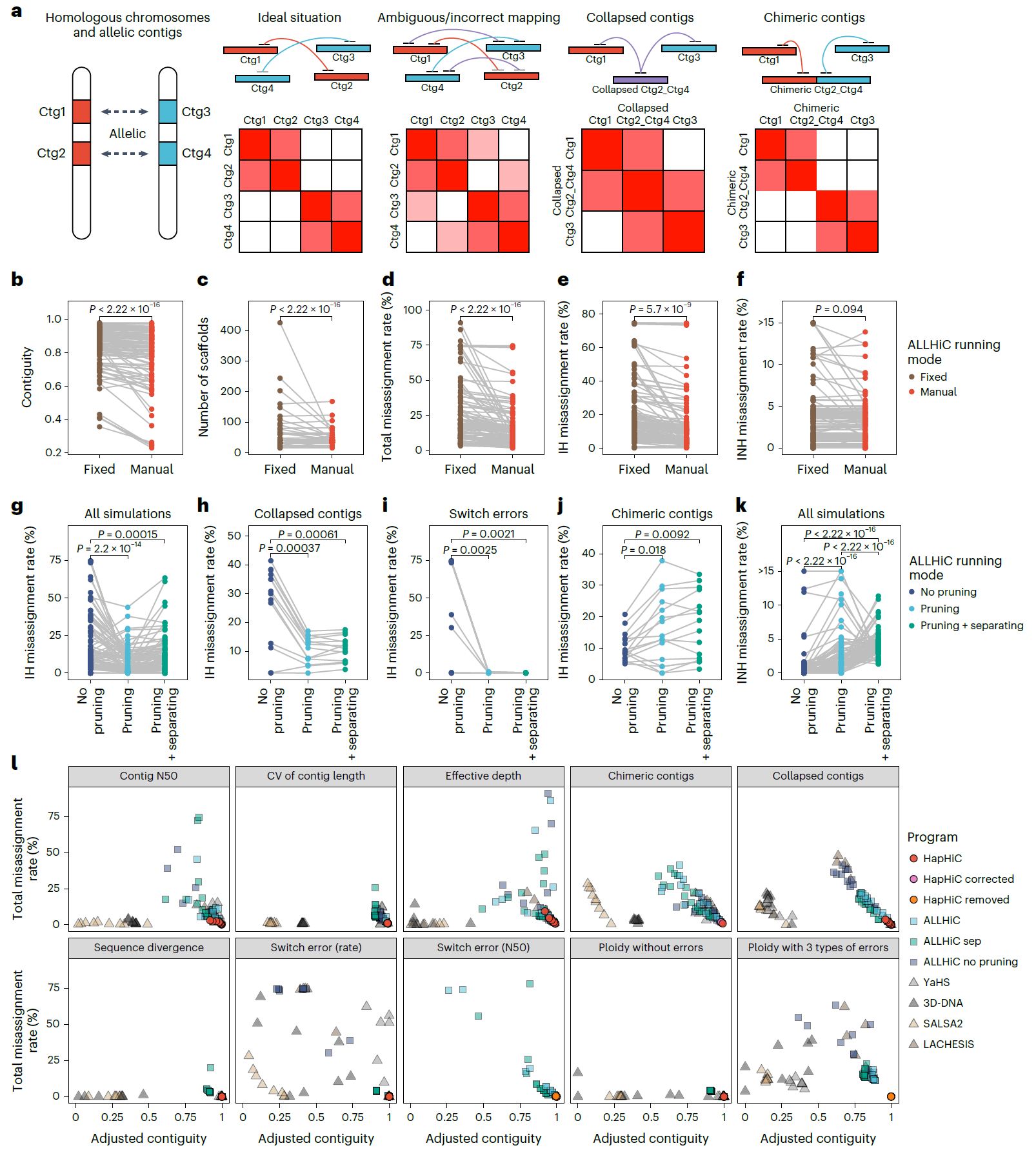
Figure 2. Comprehensive performance analysis of Hi-C-based scaffolding tools in chromosome assignment under various adverse conditions
Compared to other Hi-C scaffolding tools, HapHiC excels in both running speed and memory efficiency, significantly surpassing ALLHiC and 3D-DNA. This suggests that HapHiC offers substantial advantages in tackling the challenges of assembling large and complex genomes. Moreover, HapHiC has been successfully validated in scaffolding representative genomes of various ploidies across different taxa, including higher plants, humans, birds, amphibians, fish, insects, mollusks, and annelids. Finally, with the help of HapHiC, they constructed the haplotype-resolved allotriploid genome for Miscanthus × giganteus, an important lignocellulosic bioenergy crop.
Senior Researcher Xiaofei Zeng in Associate Professor Guoan Chen’s research team (currently an Associate Research Professor at the Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences) is the first author and one of the co-corresponding authors of the paper. Associate Professor Guoan Chen is the other co-corresponding author, and SUSTech is the first affiliated unit. Other affiliations include the Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, and Hunan Agricultural University.
This work was supported by the National Natural Science Foundation of China, Fellowship of China Postdoctoral Science Foundation, and the Science, Technology and Innovation Bureau of Shenzhen Municipality.
Related links:
Paper link in Nature Plants: https://www.nature.com/articles/s41477-024-01755-3
Research Briefing in Nature Plants: https://www.nature.com/articles/s41477-024-01756-2
HapHiC: https://github.com/zengxiaofei/HapHiC
To read all stories about SUSTech science, subscribe to the monthly SUSTech Newsletter.
Proofread ByAdrian Cremin, Yingying XIA
Photo ByDepartment of Human Cell Biology and Genetics






