Recently, the team led by Associate Professor Bo ZHANG from the Department of Biomedical Engineering at Southern University of Science and Technology (SUSTech) published a research article titled “Chemistry-Informed Deep Learning Model for Predicting Stereoselectivity and Absolute Configuration in Asymmetric Hydrogenation” in Nature Computational Science. Building on the mechanistic understanding of olefin asymmetric hydrogenation, the researchers integrated chemical knowledge into a deep-learning framework and developed a predictive model termed the Chemistry-Informed Asymmetric Hydrogenation Network (ChemAHNet). For the first time, this model enables simultaneous and accurate prediction of both the absolute configuration and stereoselectivity in the asymmetric hydrogenation of olefins bearing dual prochiral centers, representing a significant milestone for the application of artificial intelligence in complex asymmetric catalysis.
Asymmetric hydrogenation of olefins is one of the most pivotal methodologies in modern chiral synthesis, owing to its high efficiency, excellent atom economy, and broad applicability in the pharmaceutical industry. Since its recognition by the Nobel Prize in 2001, this transformation has become a core strategy for constructing enantioenriched drug molecules, significantly reducing experimental costs and accelerating drug development. However, its reliance on high-pressure hydrogen and the need to screen structurally diverse catalysts render safety and reaction-condition optimization highly challenging, underscoring the urgent demand for efficient predictive tools to guide this key process in pharmaceutical synthesis. Despite the growing use of machine learning and artificial intelligence in organic reaction prediction in recent years, their applications in asymmetric catalysis have remained significantly limited. Existing models are typically trained on relatively simple substrates containing only a single prochiral center, making it difficult for them to accurately address the more challenging asymmetric hydrogenation of olefins bearing two prochiral sites. Moreover, traditional models often focus solely on predicting enantioselectivity and are unable to simultaneously provide the absolute configuration of the major product, which is essential for the precise synthesis of complex chiral molecules. More critically, most current models rely heavily on expert-defined molecular descriptors or handcrafted features, preventing the models from autonomously learning deeper chemical principles directly from raw molecular structures. This not only restricts the upper bound of predictive performance but also severely limits the generalizability of these models to new reaction systems.
To address the practical demands of asymmetric catalysis prediction, the team developed a novel stereochemical prediction model that learns directly from molecular structures. This model eliminates the reliance on predefined molecular descriptors and can autonomously extract key reaction features from raw molecular geometries and electronic structures. As a result, it accurately predicts the outcomes of asymmetric hydrogenation reactions across diverse substrate–catalyst combinations, including olefins bearing two prochiral centers. In addition to simultaneously delivering both stereoselectivity and the absolute configuration of the major product, the model exhibits outstanding generalization capability, making it applicable to a broad range of catalytic systems and substrate classes.
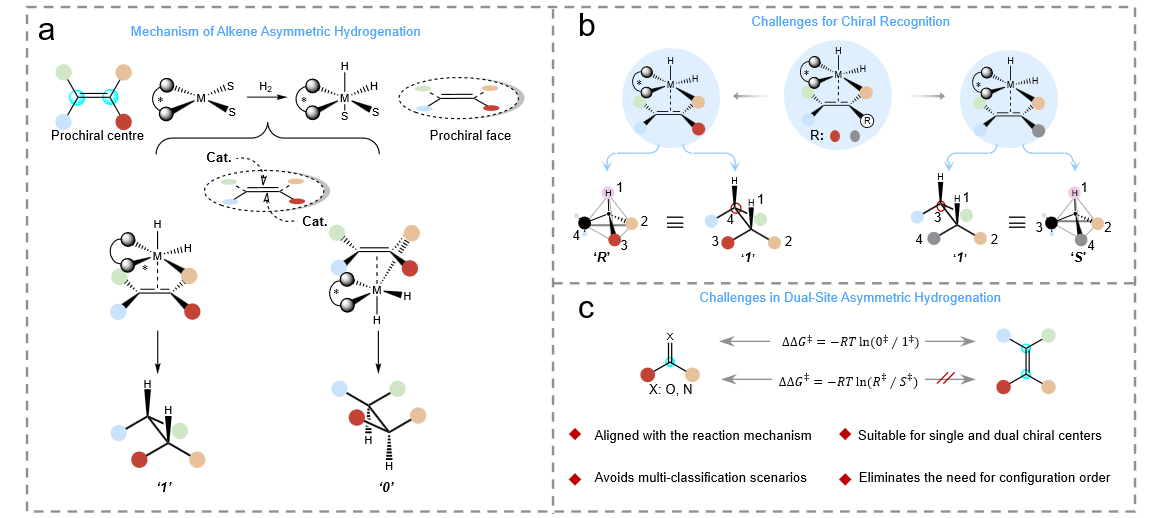
Figure 1. A Rational ∆∆G‡ Calculation Strategy for Dual-Chiral-Center Systems Based on Interaction Modes
Based on mechanistic insights into asymmetric hydrogenation and previous theoretical studies, the research team found that the carbon–carbon double bond of the olefin substrate, together with its surrounding substituents, engages in various secondary interactions with the metal center before hydrogenation (Fig. 1a). Building on this key understanding, the team proposed that the correct interaction mode between the olefin molecule and the catalytic metal center should be explicitly identified and learned within the model to fully capture the true reaction mechanism. Compared with traditional approaches that rely on the CIP rules to determine configurations, the interaction-mode framework ensures consistent predictions under the same reaction mechanism and avoids ambiguities arising from changes in substituent priorities (Fig. 1b). Moreover, for olefins bearing two prochiral centers, the interaction mode effectively overcomes the classification complexity associated with the conventional R/S system—including combinations such as (R, R), (S, S), (R, S), and (S, R)—allowing the model to accurately identify the key factors governing stereocontrol (Fig. 1c). This mechanistic rationale establishes a robust methodological foundation for accurately predicting the stereoselectivity of olefin asymmetric hydrogenation.
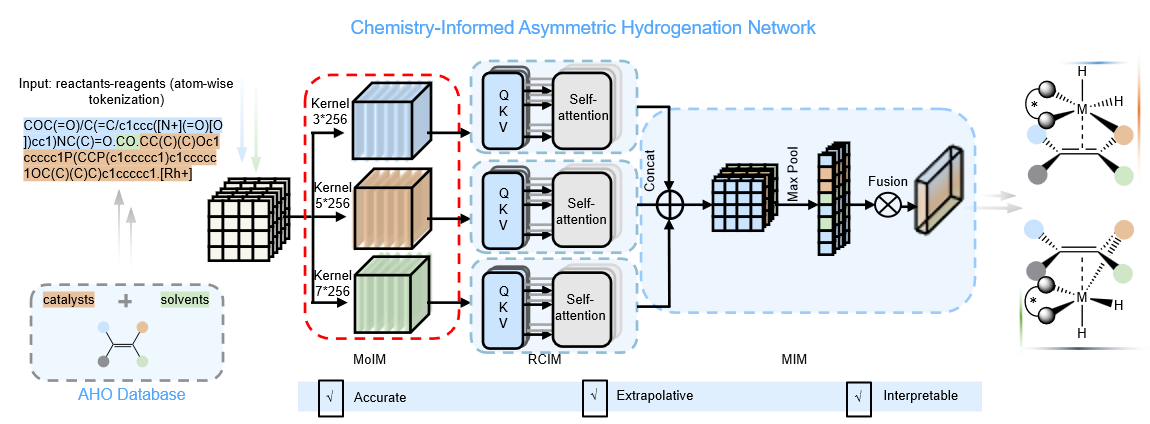
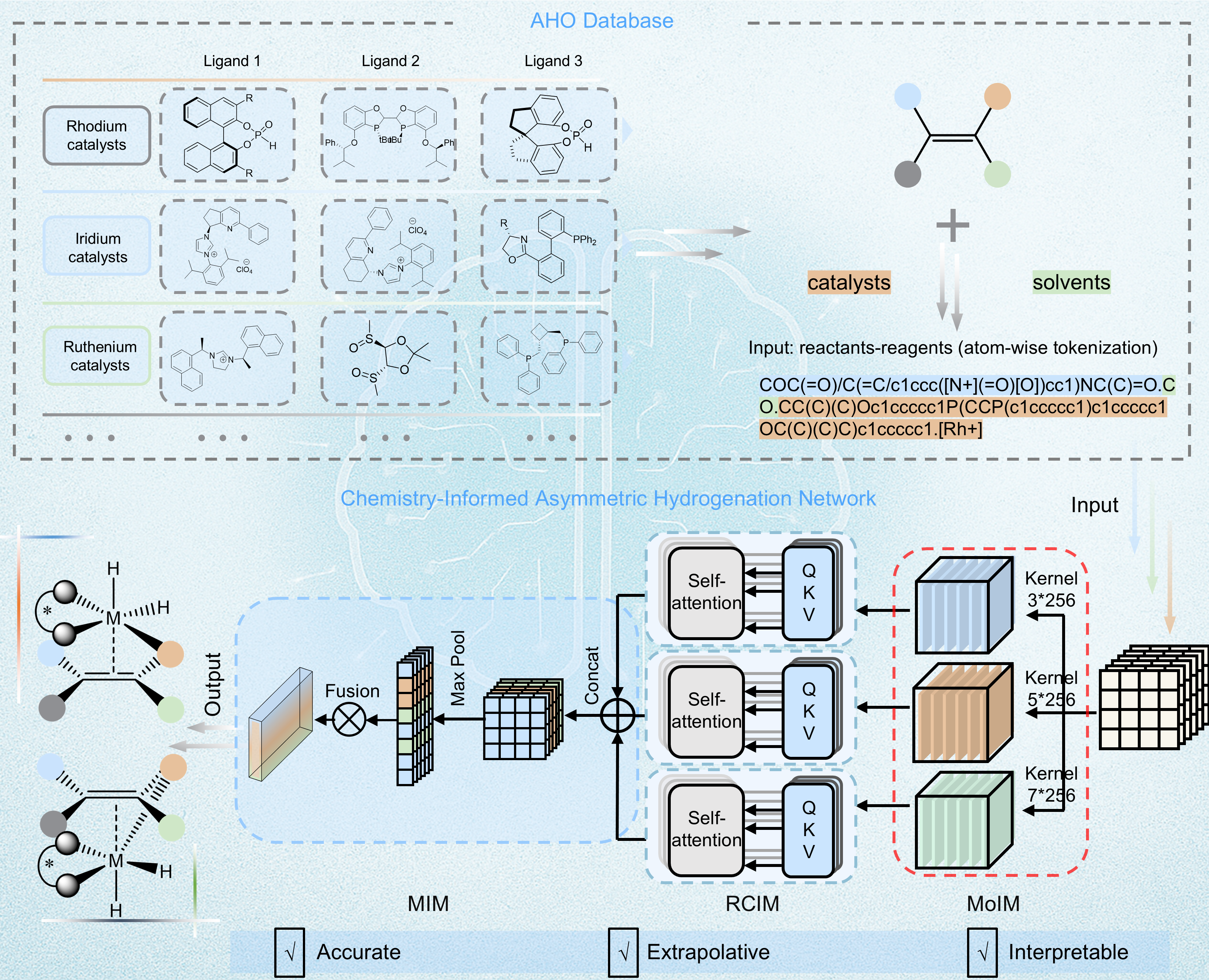
Figure 2. Conceptual Framework of the ChemAHNet Model
Inspired by these mechanistic insights, the team constructed a reaction-performance prediction model consisting of three innovative modules (Fig. 2). The Moiety Identification Module (MoIM) automatically recognizes structural moieties of different scales within each reactant and accurately captures key chemical fragments contributed by different reagents. The Reaction Components Integration Module (RCIM) integrates multi-scale moiety information from all reaction components to generate chemically meaningful molecular representations. The Molecular Interaction Module (MIM) further learns the cooperative interaction patterns among reaction components, enabling accurate identification of the true interaction mode between the olefin substrate and the metal catalytic center, thereby substantially improving the prediction of both stereoselectivity and absolute configuration. Built upon this architecture, the team developed ChemAHNet, the first model that simultaneously achieves descriptor-free learning, broad substrate applicability, absolute configuration prediction, and stereoselectivity prediction for asymmetric hydrogenation reactions.
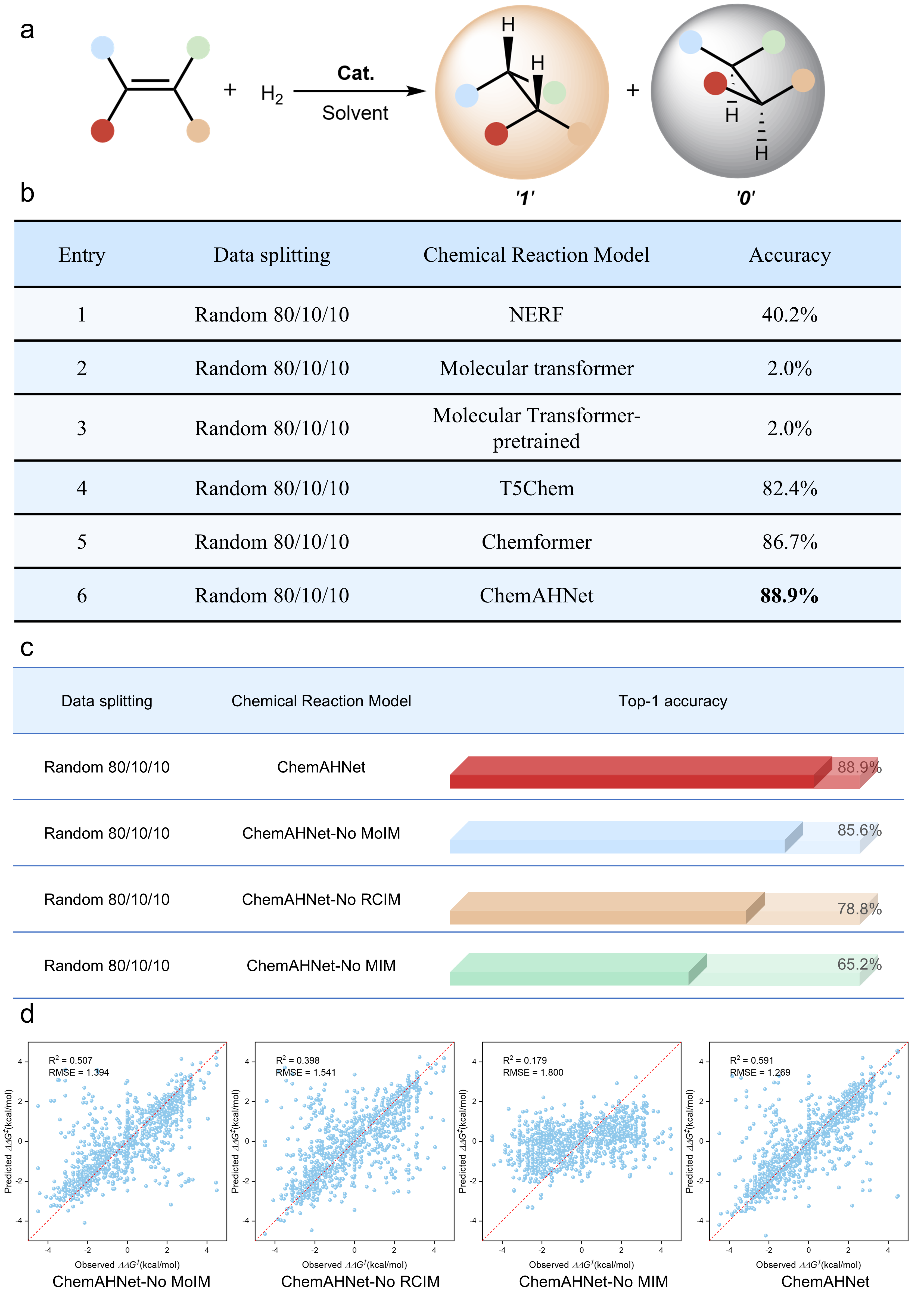
Figure 3. Performance Evaluation of the ChemAHNet Model
The results show that ChemAHNet achieves a top-1 accuracy of 88.9% in predicting the absolute configuration of the major product in olefin asymmetric hydrogenation, markedly outperforming all baseline models (Fig. 3b). This demonstrates that ChemAHNet can accurately capture the interaction mode between the substrate and the catalyst, thereby enabling effective prediction of the absolute configuration of the major enantiomer and significantly enhancing the model’s understanding of the stereocontrol principles governing olefin asymmetric hydrogenation.
Moreover, the ablation studies further confirm the necessity of the model architecture: removing any one of the MoIM, RCIM, or MIM modules leads to a noticeable drop in prediction accuracy (Fig. 3c,d). These results clearly indicate that each module plays a critical role in the overall performance of the model and that their synergistic integration is essential for achieving high-precision stereoselectivity prediction.
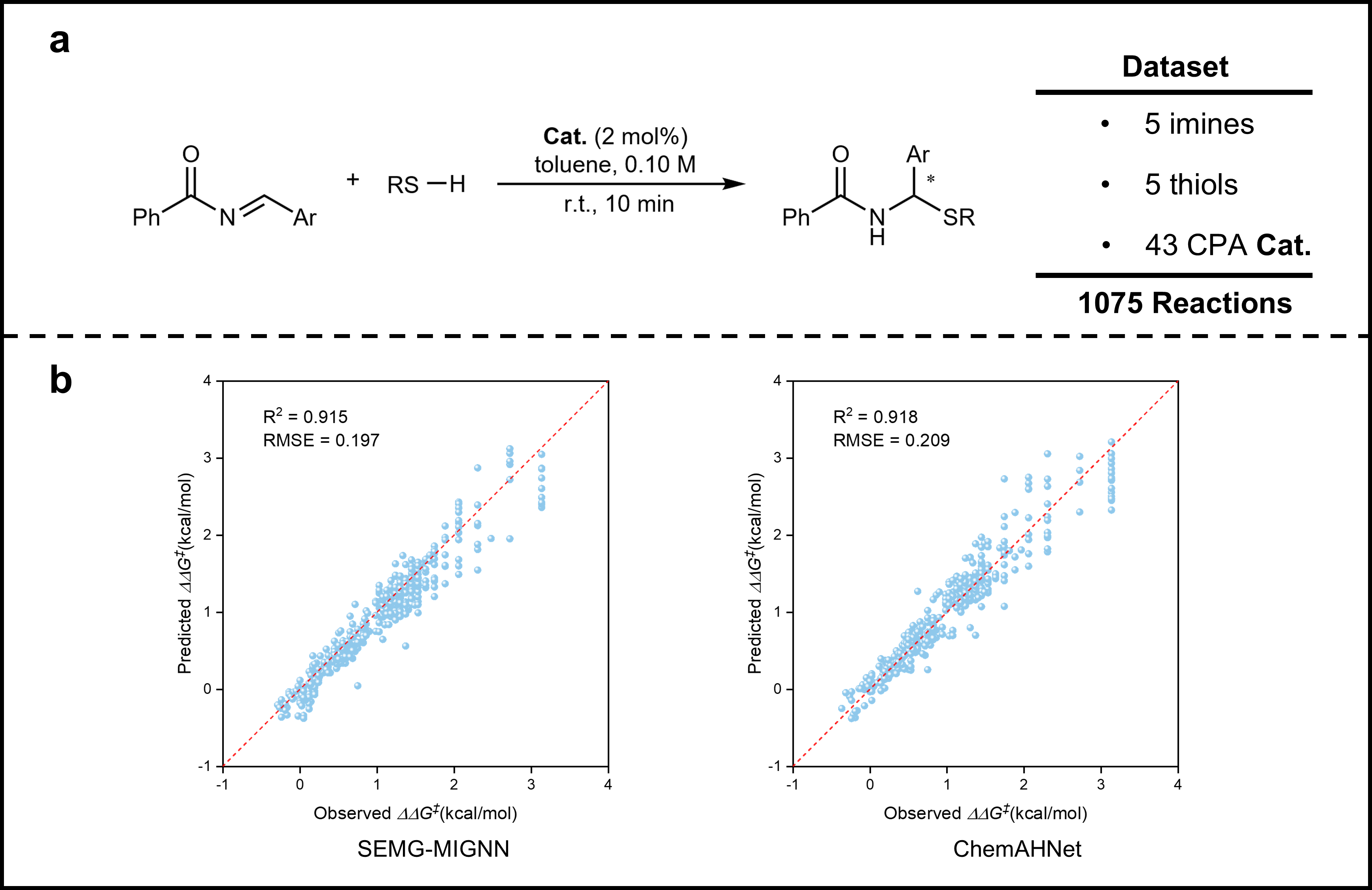
Figure 4. Predicted Enantioselectivity of the Chiral Phosphoric Acid–Catalyzed Thiol Addition to N-Acylimides
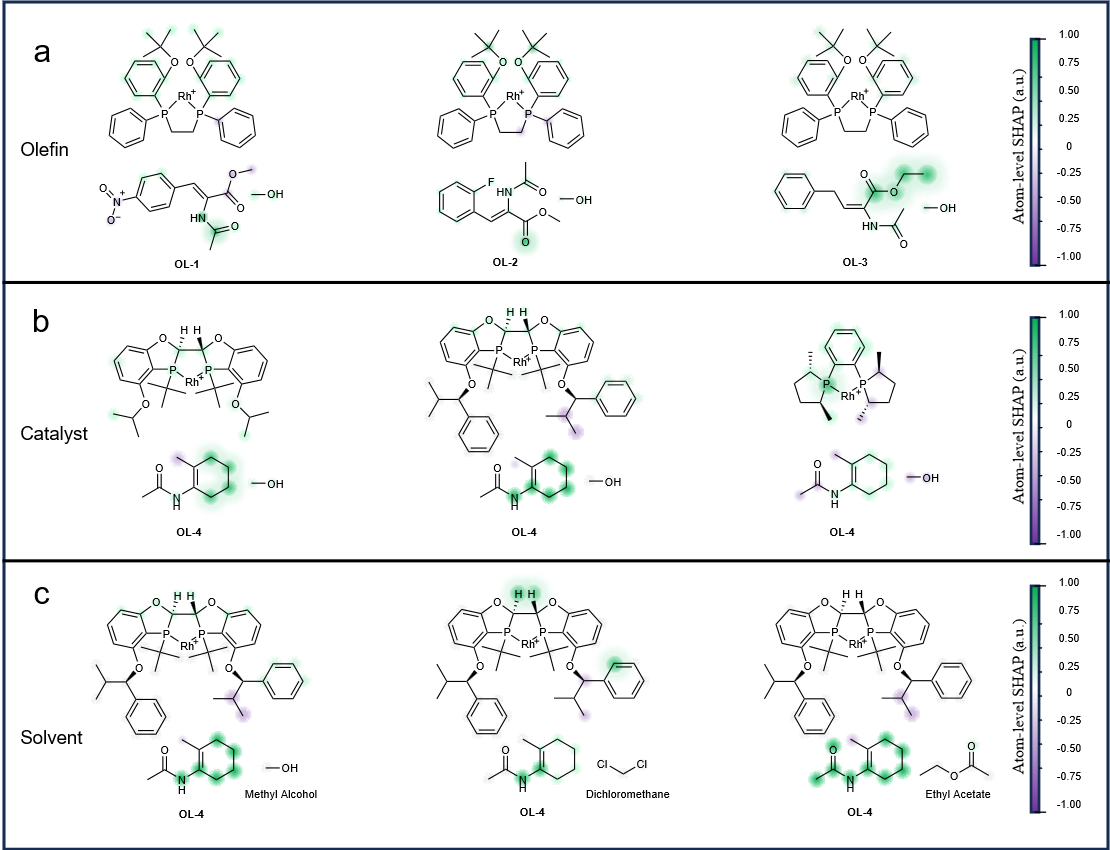
Figure 5. Chemical Interpretability of the ChemAHNet Model
To further evaluate the extensibility of ChemAHNet, we employed an external dataset involving the chiral phosphoric acid–catalyzed thiol addition to N-acylimides. The results show that ChemAHNet exhibits excellent predictive performance for the enantioselectivity of this reaction as well (Fig. 4), demonstrating that the method is not only applicable to olefin asymmetric hydrogenation but can also be effectively generalized to other types of asymmetric catalytic systems, thereby exhibiting strong potential for cross-reaction transferability. In addition, ChemAHNet provides atom-level insights into spatial and electronic interactions (Fig. 5), helping to elucidate key structural factors that govern stereoselectivity. This level of interpretability enhances the credibility of the model’s predictions and offers a more theoretically grounded and practically valuable tool for target-oriented molecular design and catalyst optimization.
The co–first authors of the paper are Li CHENG, a joint Ph.D. student (admitted in 2022) at SUSTech and the University of Macau, and Professor Panlin SHAO from Guangzhou Medical University. The corresponding authors are Bo ZHANG (SUSTech), Professor Guichuan XING (University of Macau), and Professor Panlin SHAO (Guangzhou Medical University). SUSTech serves as the leading affiliation of this work, with the University of Macau and Guangzhou Medical University as collaborating institutions.
Proofread ByNoah Crockett, Yuwen ZENG
Photo ByYan QIU






